




Leki wycofane, komunikaty bezpieczeństwa
Imbruvica (ibrutynib): Nowe srodki minimalizacji ryzyka, w tym zalecenia dotyczace modyfikacji dawki, ze wzgledu na zwiekszone ryzyko wystapienia ciezkich zdarzen sercowych

Janssen-Cilag International NV w porozumieniu z Europejskq Agencją Leków oraz Urzędem Rejestracji Produktów Leczniczych, Wyrobów Medycznych Produktów Biobójczych, przekazuje nastepujace informacje:
Preparaty Imbruvica zawierające substancję czynną Ibrutynib - zobacz w LekInfo24 »Komunikat do fachowych pracowników ochrony zdrowia
Podsumowanie
- Ibrutynib zwiększa ryzyko wystąpienia śmiertelnych i ciężkich zaburzeń rytmu serca oraz niewydolności serca.
- Pacjenci w zaawansowanym wieku, ze stanem sprawności Eastern Cooperative Oncology Group (ECOG) 2. lub współistniejącymi chorobami serca mogą być narażeni na większe ryzyko wystąpienia zdarzeń sercowych, w tym nagłych zdarzeń sercowych ze skutkiem śmiertelnym.
- Przed rozpoczęciem stosowania ibrutynibu należy dokonać odpowiedniej oceny klinicznej historii choroby i czynności serca.
- U pacjentów z czynnikami ryzyka zdarzeń sercowych, należy ocenie stosunek korzyści do ryzyka przed rozpoczęciem leczenia produktem leczniczym Imbruvica; można rozważyć zastosowanie alternatywnego leczenia.
- Pacjenci podczas leczenia powinni być dokładnie monitorowani pod kątem objawów pogorszenia czynności serca i jeśli one wystąpią, odpowiednio leczeni.
- Należy wstrzymać podawanie ibrutynibu w przypadku nowego wystąpienia lub pogorszenia się niewydolności serca stopnia 2. lub zaburzeń rytmu serca stopnia 3. Leczenie można wznowić zgodnie z nowymi zaleceniami dotyczącymi modyfikacji dawki (szczegóły poniżej).
Podstawowe informacje
Ibrutynib jest wskazany:
- w monoterapii do leczenia dorosłych pacjentów z nawracającym lub opornym na leczenie chłoniakiem z komórek płaszcza (ang. mantle cell lymphoma, MCL).
- w monoterapii lub w skojarzeniu z rytuksymabem, lub obinutuzumabem, lub wenetoklaksem, do leczenia dorosłych pacjentów z wcześniej nieleczoną przewlekłą białaczką limfocytowa (ang. chronic lymphocytic leukaemia, CLL).
- w monoterapii lub w skojarzeniu z bendamustynq_ i rytuksymabem do leczenia dorosłych pacjentów z CLL, którzy otrzymali co najmniej jedną wcześniejszą terapię.
- w monoterapii do leczenia dorosłych pacjentów z makroglobulinemią Waldenstroma (WM), którzy otrzymali co najmniej jedną wcześniejszą terapię lub pacjentów leczonych po raz pierwszy, u których nie jest odpowiednie zastosowanie chemioimmunoterapii. Ibrutynib w skojarzeniu z rytuksymabem jest wskazany do leczenia dorosłych pacjentów z WM.
Ocena danych ze zbioru randomizowanych badań klinicznych ibrutynibu wykazała niemal 5-krotnie większą częstość występowania zdarzeń nagłego zgonu sercowego, nagłego zgonu lub zgonu sercowego w ramieniu ibrutynibu (11 przypadków; 0,48%) w porównaniu z ramieniem komparatora (2 przypadki; 0,10%). Po skorygowaniu o ekspozycje, zaobserwowano 2-krotny wzrost wskaźnika częstości występowania (EAIR, wyrażony jako liczba uczestników ze zdarzeniami podzielona przez liczbę pacjento-miesięcy pozostających w narażeniu) zdarzeń nagłego zgonu sercowego, nagłego zgonu lub zgonu sercowego w ramieniu ibrutynibu (0,0002) w porównaniu z ramieniem komparatora (0,0001).
Na podstawie oceny dostępnych danych dotyczących kardiotoksyczności ibrutynibu, do druków informacyjnych produktu leczniczego wprowadzono dalsze środki mające na celu zminimalizowanie ryzyka kardiologicznego. Pacjenci w zaawansowanym wieku, w stanie sprawności >2. wg Eastern Cooperative Oncology Group (ECOG) lub ze współistniejącymi chorobami serca mogą być bardziej narażeni na ryzyko wystąpienia zdarzeń, w tym nagłych zdarzeń sercowych ze skutkiem śmiertelnym.
Przed rozpoczęciem stosowania produktu leczniczego Imbruvica należy przeprowadzić odpowiednia ocenę kliniczną wywiadu i czynności serca. Pacjenci powinni być uważnie monitorowani w trakcie leczenia, w celu wykrycia objawów klinicznego pogorszenia czynności serca i jeśli one wystąpią, powinni być odpowiednio leczeni. U pacjentów, u których istnieją zagrożenia sercowo-naczyniowe, należy rozważyć dalsza ocenę (np. EKG, echokardiogram), jeśli jest to wskazane.
U pacjentów z istotnymi czynnikami ryzyka zdarzeń sercowych, przed rozpoczęciem leczenia produktem leczniczym Imbruvica należy starannie ocenić stosunek korzyści do ryzyka; można rozważyć zastosowanie alternatywnego leczenia.
Punkt 4.4 ChPL (Charakterystyki Produktu Leczniczego) został odpowiednio zaktualizowany, a w punkcie 4.8 ChPL dodano zatrzymanie krążenia jako działanie niepożądane patrz: https ://www.janssen.com/poland/produkty.
Ponadto, podmiot odpowiedzialny przeanalizował dane kliniczne pacjentów doświadczających zdarzeń sercowych stopnia 3.+ i ocenił czy toksyczności nawróciły u pacjentów, u których zmniejszono dawkę produktu leczniczego Imbruvica, w porównaniu z pacjentami, u których nie zmniejszono dawki w następstwie tych toksyczności. Analizy wskazują na mniejsza częstość występowania nawrotów zdarzeń sercowych u pacjentów, u których zmniejszono dawkę produktu leczniczego Imbruvica, w porównaniu z osobami, u których nie zmniejszono dawki produktu leczniczego Imbruvica.
Na tej podstawie, punkt 4.2 ChPL został zaktualizowany o następujące nowe zalecenia:
Należy przerwać stosowanie produktu leczniczego Imbruvica w razie nowego wystąpienia lub nasilenia: niewydolności serca stopnia 2., zaburzeń rytmu serca stopnia 3. Gdy objawy toksyczności zmniejsza się do stopnia 1. lub ustąpią, można wznowić leczenie produktem leczniczym Imbruvica w zalecanej dawce zgodnie z poniższą tabelą:
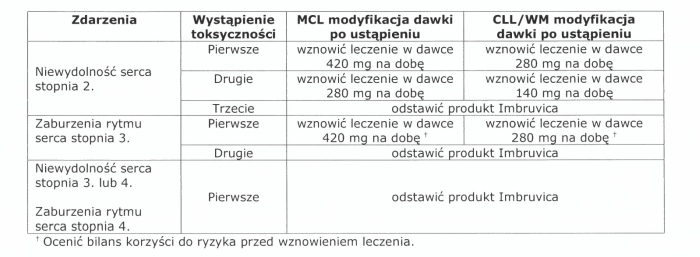
Zalecane modyfikacje dawki w przypadku zdarzeń niekardiologicznych (toksyczność niehematologiczna stopnia >3., neutropenia stopnia >3. z zakażeniem lub gorączką, lub toksyczność hematologiczna stopnia 4.) pozostają w większości bez zmian, z dodaniem jedynie następującego przypisu w tabeli: „Przy wznowieniu leczenia należy ponownie rozpocząć je od tej samej lub mniejszej dawki w oparciu o ocenę korzyści i ryzyka. W przypadku ponownego wystąpienia toksyczności należy zmniejszyć dawkę dobowa o 140 mg".
Zgłaszanie działań niepożądanych
Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane związane ze stosowaniem produktu Imbruvica zgodnie z zasadami zgłaszania działań niepożądanych za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych:
Al. Jerozolimskie 181C
02-222 Warszawa
Tel.: + 48 22 49 21 301
Faks: + 48 22 49 21 309
Strona internetowa: https://smz.ezdrowie.gov.pl;
lub bezpośrednio do podmiotu odpowiedzialnego.
Dane kontaktowe podmiotu odpowiedzialnego
Janssen-Cilag Polska Sp. z o.o.
ul. Iłżecka 24
02-135 Warszawa
JanssenPVPoland@its.jnj.com
tel.: +48 22 237 60 00
Powiązane produkty
Reklama

Reklama
Reklama
Reklama
Reklama
Reklama
Reklama

